overview¶
Options¶
Option |
Description |
Argument |
|---|---|---|
--samples |
Sample names. It determines the plotting order If “NA” all samples are used [default NA] |
[char …] |
--gipOut |
GIP output directory [default gipOut] |
[char] |
--outName |
Output name [default gipOut/sampleComparison/overview] |
[char] |
--chrs |
Chromosomes to use. If “NA” it uses the same chromsomes as GIP [default NA] |
[char …] |
--regions |
Bed3 file with regions of interest. Regions must be included in
|
[chr] |
--MAPQ |
Label bins with MAPQ < –MAPQ [default 0] |
[int] |
--ploidy |
Genome ploidy. After normalization multiply coverage values by –ploidy [default 2] |
[int] |
--highLowCovThresh |
Provide two numbers. Bins with normalized coverage values > num1 or < num2 will be labeled [default 1.5 0.5] |
[double] |
--minDelta |
Remove genes with normalized coverage delta < –minDelta [default 0] |
[double] |
--ylim |
Plot visualization threshold. Bin or gene normalized coverage values > –ylim are shown as –ylim [default 5] |
[double] |
--ylimInt |
Bin scatterplot y-axis values interval. If "NA" it is automatically assigned [default NA] |
[double] |
--maxGe |
Plot visualization threshold. Max number of genes to show (ordered by delta coverage) [default 10000] |
[int] |
--binPlotDim |
Bin coverage plot height and width values [default 10 20] |
[double double] |
--gePlotDim |
Gene coverage plot height and width values [default 10 20] |
[double double] |
--chrPlotDim |
Chromosome coverage plot height and width values [default 10 20] |
[double double] |
--regPlotDim |
Regions of interest plot height and width values [default 10 20] |
[double double] |
--noBin |
Omit bin coverage output |
|
--debug |
Dump session and quit |
|
-h, --help |
Show help message |
Description¶
overview module aims at comparing the samples in terms of chromosomes, genomic bins and genes sequencing coverage. Unlike other modules like binCNV or geCNV where normalization accounts for chromosome copy number, data in overview is normalized by median genome coverage only. overview is suited to display CNV variation in multiple samples with respect to the reference genome. overview does not perform sequencing coverage ratios between samples. The normalized scores do not represent somy scores. The normalization procedure is such that the coverage of most genomic bins will be centered on 1. The --ploidy parameter can be used to indicate the organism ploidy level, which is 2 by default (diploid). overview multiplies the normalized coverage scores of bins, genes and chromosomes by the ploidy value.Example¶
giptools overviewThe .chrCov.pdf file represents the normalized chromosome coverage
The .binCov.pdf file represents the normalized genomic bin coverage
The .geCov.pdf file represents the normalized gene coverage. The first heatmap reports scaled values. Genes with 0 standard deviation cannot be scaled and are not shown in the hetmap. The second heatmap shows all the normalized gene coverages increased by a pseudocount of 0.1 and log2 transformed. The third heatmap shows the actual normalized gene coverage, but values greather than –ylim are reported as –ylim.
The .geCov.xlsx file is an excel table reporting the normalized gene coverage with the associated function (if available)
The .alignmentMetrics.xlsx is an excel table reporting the mapping statistics of all samples as estimated by picard CollectAlignmentSummaryMetrics.
--ploidy 1 --ylim 2.5 --highLowCovThresh 100 -1 to the command. This gives the following plot: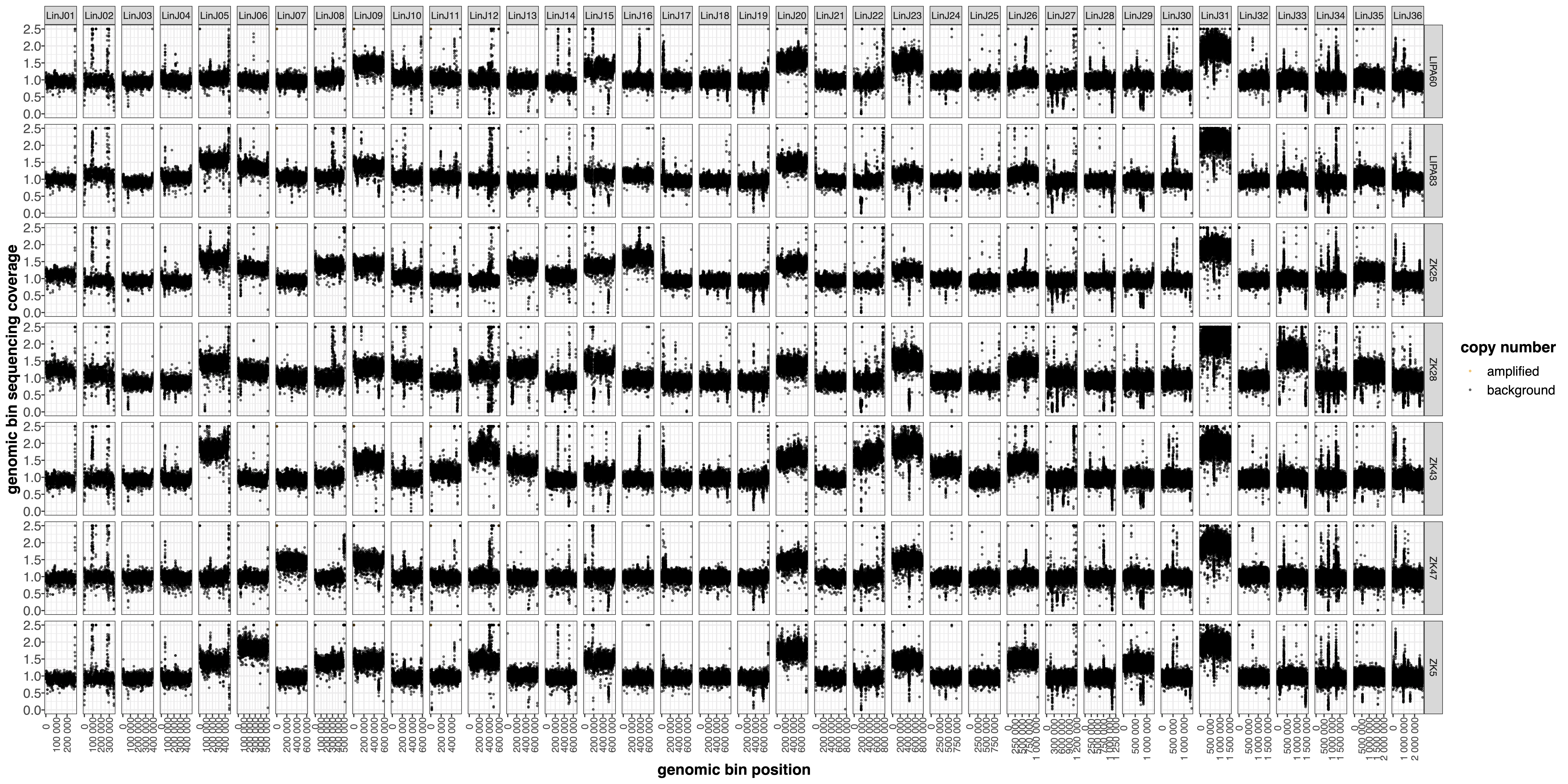
--highLowCovThresh 1.25 0.5 --MAPQ 50 can be used to color the genomic bins with normalized coverage above 1.25 and to label low MAPQ bins: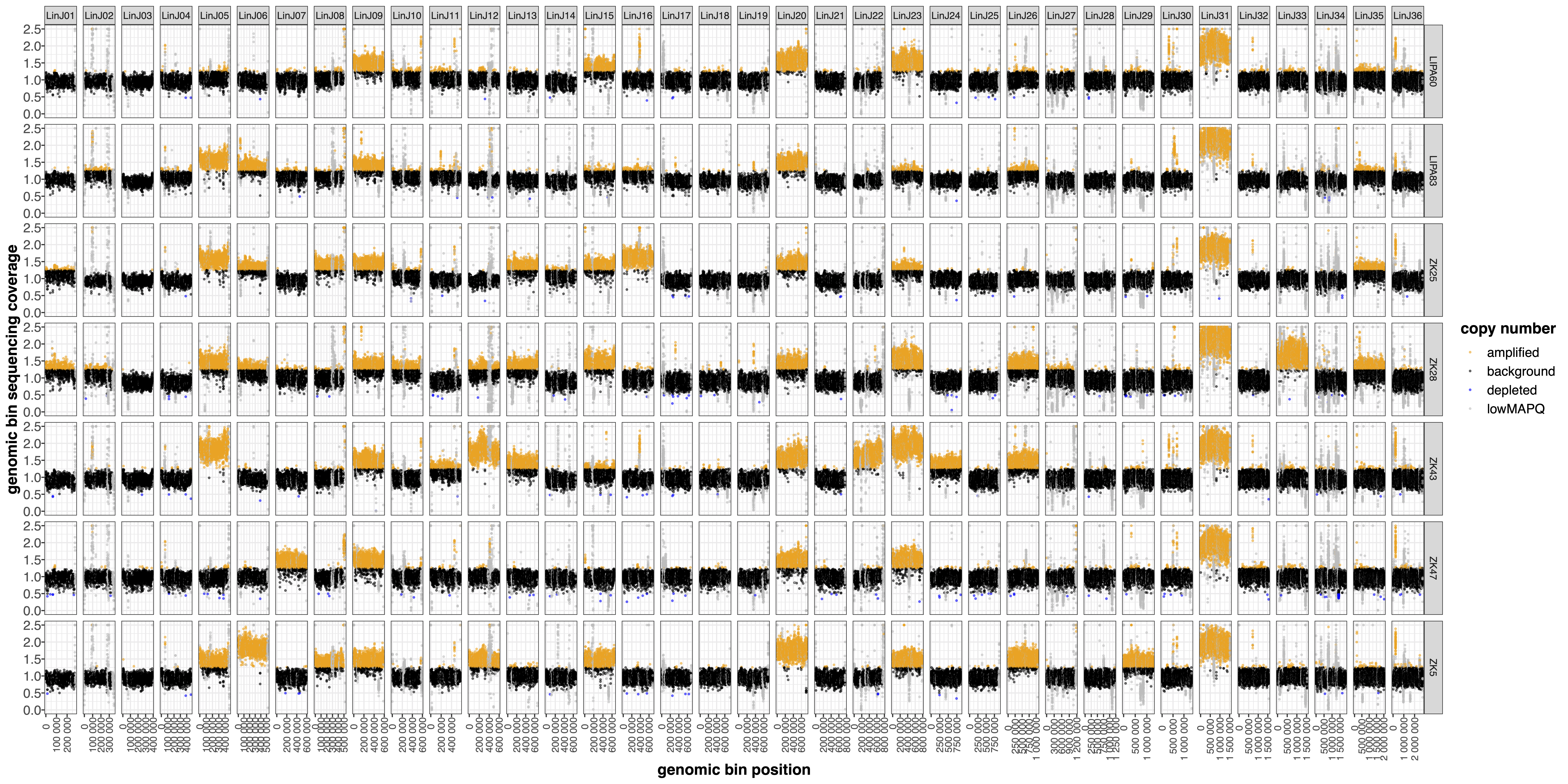
regions parameter file with genomic coordinates in Bed3 format (i.e. chromosome<Tab>start<Tab>end), the user can zoom on specific regions of interest (.binCovRegions.pdf output)