geCNV¶
Options¶
Option |
Description |
Argument |
|---|---|---|
--samples |
Two sample names. The ratio is computed Sample2/Sample1 [required] |
[char,char] |
--gipOut |
GIP output directory [default gipOut] |
[char] |
--outName |
Output name [default gipOut/sampleComparison/geCNV] |
[char] |
--chrs |
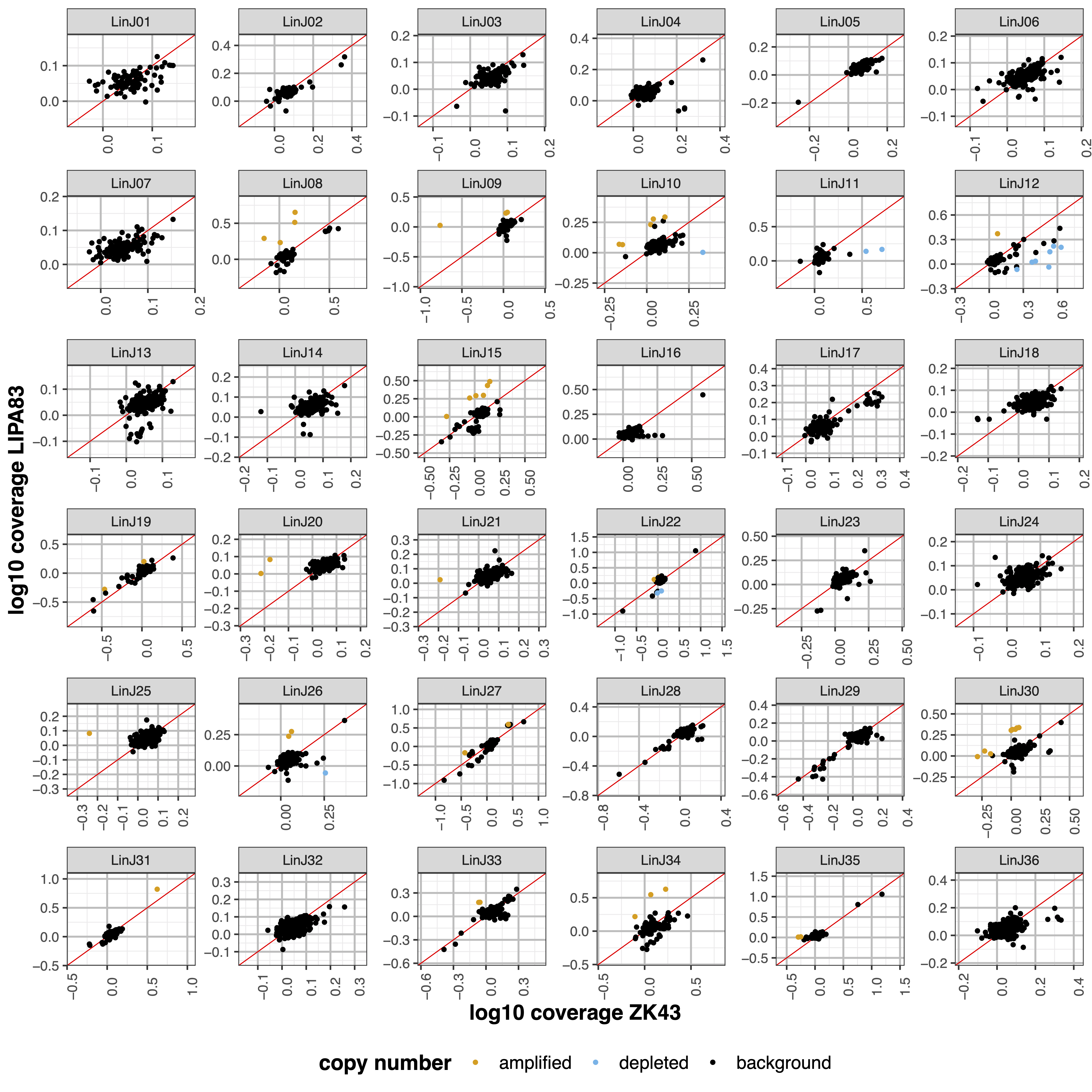
Chromosomes to use. If “NA” it uses the same chromsomes as GIP [default NA] |
[char …] |
--MAPQ |
Label genes with MAPQ < –MAPQ [default 0] |
[int] |
--highLowRatio |
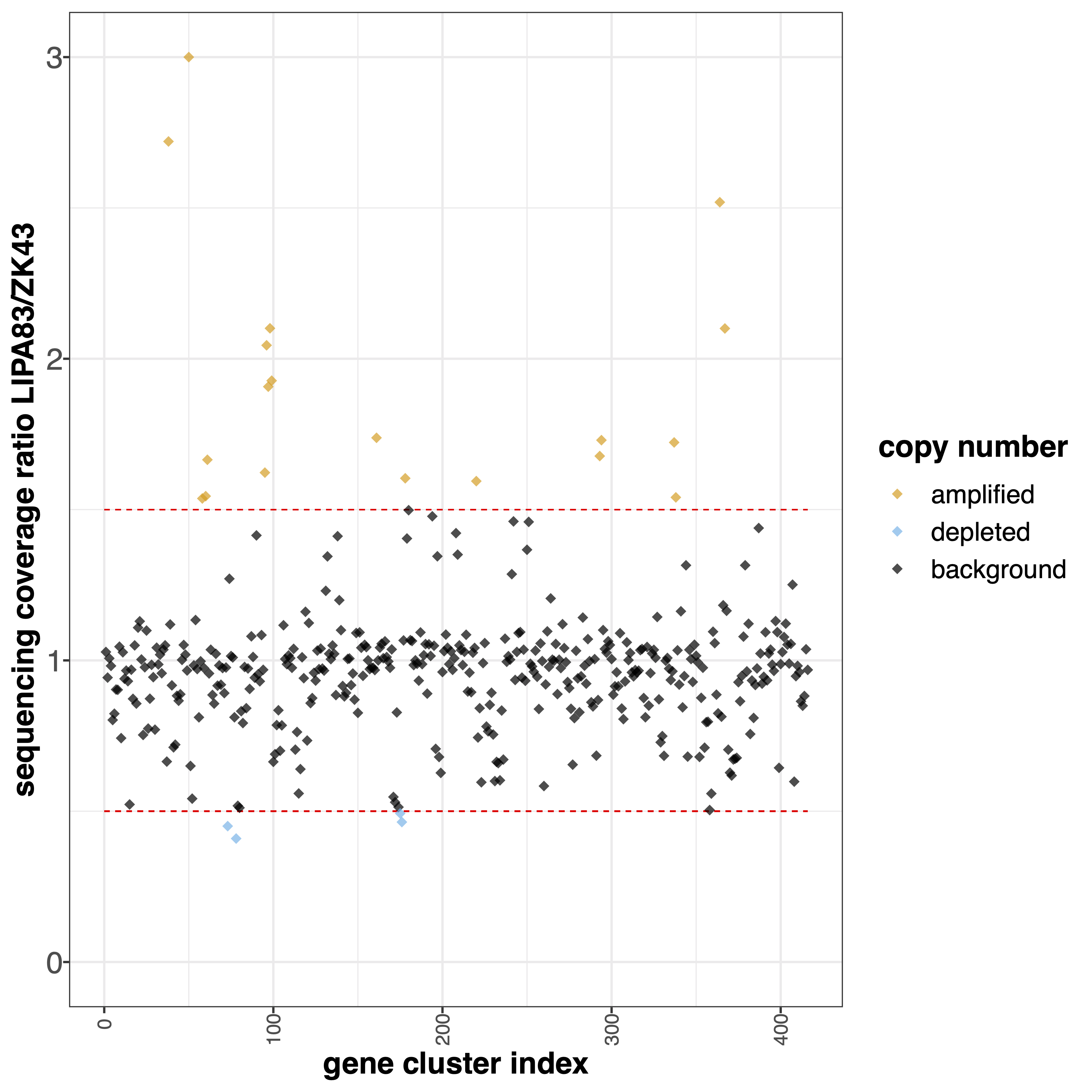
Provide 2 numbers. Genes with ratio scores > num1 or < num2 will be colored differently [default 1.5 0.5] |
[double,double] |
--pseudocount |
Normalized mean coverage pseudo count value preventing infinite (1/0) and NaN (0/0) ratio values [default 0.1] |
[double] |
--overview_ylim |
Overview plot visualization threshold. Ratio values greather than this threshold are shown as –ylim [default 3] |
[double] |
--scaleFree |
Graphical parameter multi-panel plots Set scale free axes [default yes] |
[yes|no] |
--multi_min |
Multi-panel plot 1 visualization threshold. Min normalized gene coverage DEPENDENCY:–scaleFree no [default 0] |
[double] |
--multi_max |
Multi-panel plot 1 visualization threshold. Max normalized gene coverage DEPENDENCY:–scaleFree no [default 100] |
[double] |
--multiLog_min |
Multi-panel plot 2 visualization threshold. Min normalized gene coverage (log10 scale). DEPENDENCY:–scaleFree no [default -1] |
[double] |
--multiLog_max |
Multi-panel plot 2 visualization threshold. Max normalized gene coverage (log10 scale). DEPENDENCY:–scaleFree no [default 3] |
[double] |
--debug |
Dump session and quit |
|
-h, --help |
Show help message |
Description¶
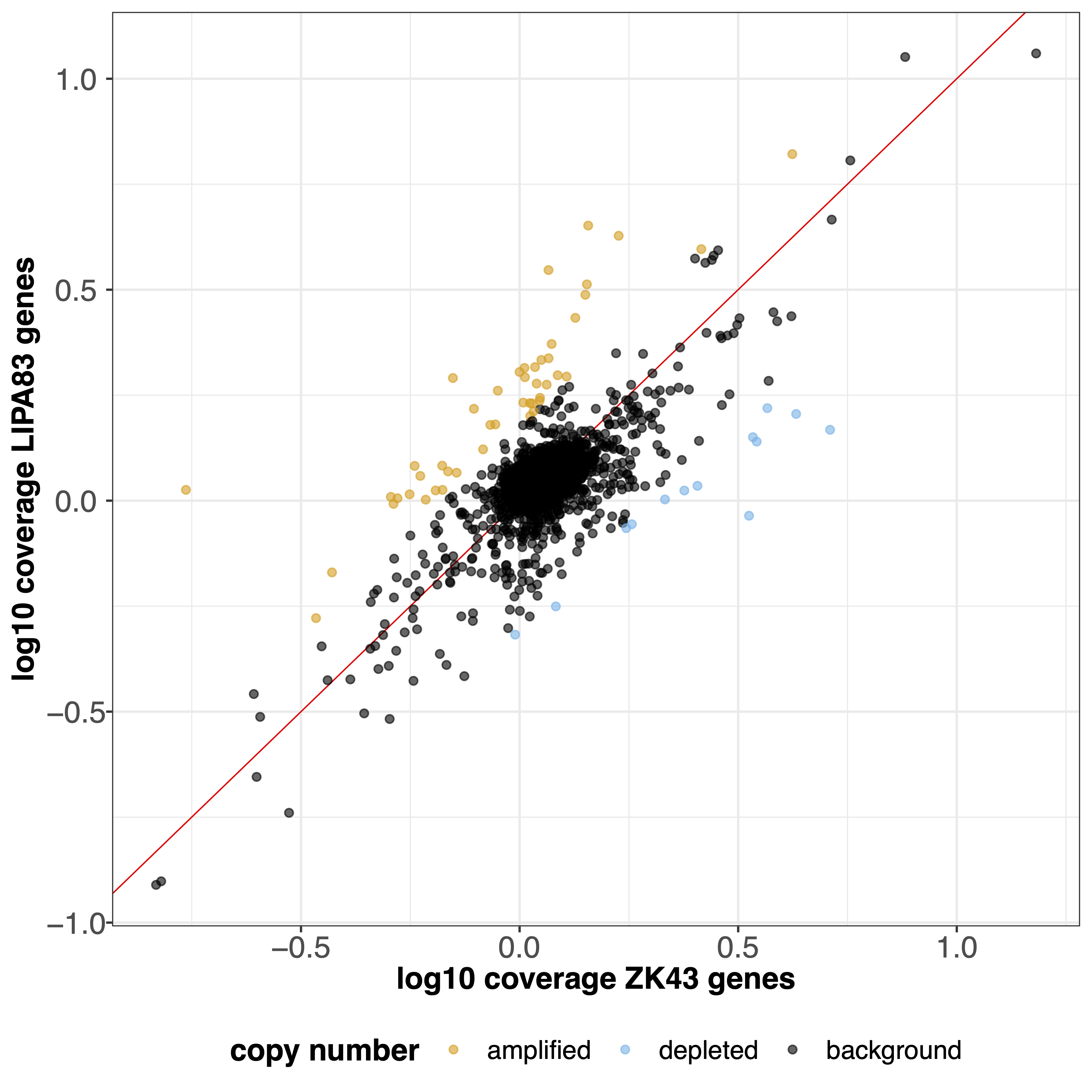
geCNV module aims at comparing the gene sequencing coverage of 2 samples to identify gene CNVs.Example¶
giptools geCNV --samples ZK43 LIPA83